Scientists speed up muscle repair—could fight dystrophy

Baltimore, MD---Athletes, the elderly and those with degenerative muscle disease would all benefit from accelerated muscle repair. When skeletal muscles, those connected to the bone, are injured, muscle stem cells wake up from a dormant state and repair the damage. When muscles age, however, stem cell number and function declines, as do both tissue function and regenerative ability. Carnegie’s Christoph Lepper and team*, including researchers from the University of Missouri, investigated muscle stem cell pool size. In particular, they asked if stem cell number could be increased, and if there would be any associated functional benefits.
Using genetically modified mice, the scientists found that while a muscle’s size remained unchanged, it surprisingly, is capable of supporting a much greater number of these stem cells than previously thought. These “super-numeral” stem cells could repair injured muscle and were faster at it than when only normal numbers are present. The team also found that the increase in stem cells stunts the decline of weakened, degenerative muscles, potentially a boon for fighting muscular dystrophy. The study is published in the October 11, 2016, issue of eLife.
Muscle stem cells, called satellite cells, are undifferentiated muscle cells that promote growth, repair and regeneration. As Lepper explained: “These satellite cells make up some 5-7% of all muscle cells and are essential to muscle regeneration. When a mouse is born, the satellite cells divide and differentiate for about 3 to 4 weeks driving tissue growth. They then go quiet until an injury is detected. The number of satellite cells set aside at this time appears to be relatively constant with regard to the host muscle tissue size. We wanted to see whether this ratio could be manipulated and, if so, whether there would be any physiological consequences.”
Lepper’s collaborator Richard Tsika had previously generated mice that overexpressed a gene called TEAD1 and found that the protein the gene produces, TEAD1, affects the regulation of the type of muscle fiber produced. The current study documents the dramatic effect TEAD1-expressing muscle fibers have on their associated satellite cells, which do not express TEAD1. TEAD1 transgenic mice have up to a six-fold increase in the number of satellite cells, which was true across all muscle groups that were analyzed.
This study facilitated the surprising discovery that the muscle fiber can “communicate” to its stem cells to influence the stem cell pool size. This molecular communication to the satellite cells was the origin of the stem cell increase.
“We were very surprised to find that it was possible to uncouple the number of stem cells from the host tissue size without seeing negative consequences to muscle physiology,” remarked Sheryl Southard, co-lead author on the paper with Ju-Ryoung Kim. Remarkably, the increased number allowed muscles to regenerate much faster after injury.
Importantly, the scientists found that in a mouse model for Duchenne muscular dystrophy, TEAD1 overexpression stunted the wasting disease.
The researchers suggest that the increase in the number of satellite cells, without any changes to overall muscle size, makes the genetically modified TEAD1 mouse a good model organism. With it, they hope to discover the molecular cascade that regulates muscle stem cell number and the “stop” and “go” signals that cause the cells to differentiate and go quiet.
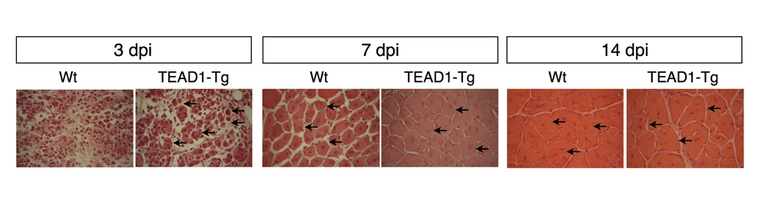
Caption: A normal muscle fibers are shown in the left panels at three, seven, and 14 days post injury. The fibers in the right panel are from the genetically modified mice that were overexpressing the TEAD1 gene, which resulted in a significant increase in muscle stem cells. The bright red dots and arrows indicated the muscle stem cells. Image courtesy Sheryl Southard.
*The authors on the paper are Sheryl Southard, Ju-Ryoung Kim, SiewHui Low (also a Carnegie researcher), Richard Tsika, and Christoph Lepper. This work was supported by NIH grant DP5OD009208, AR41464, and the Carnegie Institution for Science.